Et celles des autres années dans le menu "burger"
Et via le lien ci-dessous
E-mail ou mot de passe invalide
> Mot de passe oublié Je n'ai pas de compte Créer un compte
Quand y penser ? L’heure du bilan, l’heure du traitement.
Le terme de « syndrome myélodysplasique » (SMD) s’applique à un ensemble d’hémopathies qui se caractérisent par le déficit progressif d’une ou plusieurs catégories de cellules sanguines d’origine myéloïde (hématies, granulocytes, plaquettes) ; la moelle est paradoxalement riche mais les lignées hématopoïétiques présentent des troubles de maturation justifiant l’appellation de « dysplasie » ; il s’agit d’une dysplasie au sens cancérologique du terme puisqu’elle comporte une part monoclonale et que certains de ces états évoluent après un délai variable vers une leucémie aiguë. Les SMD comprennent en particulier le groupe des « anémies réfractaires » qui constitue un cadre diagnostique précis.
Le premier cas décrit remonte à 1900 ; la fréquence est mal connue car ce sont des maladies de la personne âgée, souvent non diagnostiquées, en particulier dans un contexte de co-morbidité :S’il existe peu de données épidémiologiques fiables mais quelques études récentes en Europe et aux Etats-Unis concluent que les SMD sont sans conteste la première cause d’hémopathie maligne après 60 ans : l’incidence augmente régulièrement avec l’âge jusqu’à concerner 1 sujet sur 6 de plus de 90 ans dans la récente cohorte américaine de Greenberg. Sans atteindre ces chiffres considérables, force est de constater une réelle inflation de cas, sans commune mesure avec l’accroissement de l’effectif des hématologues : le rôle des médecins généralistes dans la prise en charge de cette pathologie est donc appelé à s’amplifier.
La cause de la dysplasie hématopoïétique est généralement inconnue mais certains facteurs favorisants sont bien identifiés : 5% des patients ont guéri d’un précédent cancer au prix d’une exposition aux radiations ionisantes et/ou à la chimiothérapie. On retrouve par ailleurs dans 17% des cas une antériorité plus ou moins lointaine d’anémie inexpliquée ou d’autre cytopénie. Le rôle d’une sénescence du système immunitaire est probable. Certains facteurs d’environnement sont suspectés mais sans argument déterminant.
Quand y penser ?
En principe chez une personne âgée de plus de 60 ans ; 9 fois sur 10 le diagnostic est évoqué sur un résultat d’hémogramme systématique et dans 90% des cas l’anémie est le signe d’appel : elle est généralement macrocytaire. Le laboratoire signale souvent des dystrophies des cellules sanguines: anisocytose, poïkilocytose, hypogranularité ou défaut de lobulation du noyau des polynucléaires, anisothrombocytose, plaquettes géantes… Une autre cytopénie isolée est plus rare mais s’associe volontiers à l’anémie (neutropénie dans 30% des cas et thrombopénie dans 10%), et tend à apparaître ou se majorer avec l’évolution. Les SMD sont généralement des hémopathies chroniques qui restent longtemps bien supportées ; cliniquement il peut exister des signes d’insuffisance médullaire : syndrome anémique principalement mais aussi infections fréquentes ou purpura.
Le diagnostic de SMD nécessite l’exclusion par des analyses complémentaires d’un certain nombre de causes d’anémie : carences vitaminiques B9 et B12, insuffisance rénale, hypothyroïdie, maladies inflammatoires chroniques, dysglobulinémies. Devant une anémie normocytaire ou macrocytaire non régénérative, une exploration endoscopique digestive à la recherche d’un saignement est tout à fait inutile, d’autant que la ferritine sérique est souvent élevée. Un traitement d’épreuve par acide folique et vitamine B12 peut être administré pendant quelques semaines, les sels ferreux sont en revanche à éviter. En cas de thrombopénie isolée, il faudra également éliminer une carence mais surtout l’intoxication alcoolique chronique et les hépatopathies en général, s’intéresser aussi à la monocytose pouvant orienter vers une Leucémie myélo-monocytaire chronique (LMMC). Une neutropénie seule doit faire écarter une simple anomalie de répartition des leucocytes, en particulier chez les femmes, ainsi qu’une une cause toxique ou médicamenteuse.
L’heure du bilan
Hormis le cas d’une présentation pancytopénique d’emblée évocatrice, le diagnostic de SMD ne s’envisage qu’après un recul de 6 mois sur la première constatation d’une anémie ou d’une autre cytopénie. Les cytopénies modérées sans retentissement clinique doivent être simplement surveillées par un hémogramme mensuel ; en cas de tendance évolutive, d’apparition d’une symptomatologie fonctionnelle ou de pathologie associée (insuffisance cardiaque en particulier), il faut approfondir le bilan. La consultation d’hématologie spécialisée est alors nécessaire car le diagnostic et l’évaluation pronostique requièrent une expertise en cytologie sanguine et médullaire ainsi qu’une étude cytogénétique des précurseurs hématopoïétiques.
Une fois le diagnostic établi, la classification du SMD en cause est indispensable, elle permet de distinguer les formes les plus graves (30%) qui comportent une proportion excessive de blastes dans la moelle et se trouvent donc déjà sur le versant leucémique. Cette classification (OMS 2000) se base sur le nombre de lignées dysplasiques, la présence ou non d’une rétention du fer mitochondrial dans les érythroblastes (sidéroblastes en couronne), la proportion des blastes dans la moelle et le caryotype. Les catégories ainsi définies sont :
1) l’anémie réfractaire, simple ou sidéroblastique
2) l’anémie réfractaire avec délétion du bras long du chromosome 5 (syndrome 5q- )
3) les cytopénies réfractaires avec dysplasie de plusieurs lignées ± sidéroblastes
4) les anémies ou cytopénies réfractaires avec excès de blastes.
L’évaluation pronostique repose depuis 1997sur le score IPSS (International Prognostic Scoring System) dont le calcul combine 3 critères : le nombre de cytopénies sanguines, le caryotype (valeur plus ou moins péjorative de certaines anomalies) et le pourcentage de blastes médullaires. Ce score est prédictif du risque de décès et de celui de transformation leucémique aiguë : 70% des SMD sont de faible risque (médiane de survie = 8 ans) et 30% de risque élevé (médiane de survie = 18 mois).
_ Les SMD de haut risque relèvent d’une prise en charge spécialisée si l’âge le permet : le traitement s’apparente à celui des leucémies aiguës, faisant appel à des protocoles lourds de chimiothérapie, à la greffe de cellules souches hématopoïétiques si possible, aux immunosuppresseurs majeurs dans certaines cas, à l’usage des nouveaux médicaments déméthylants (azacytidine, décitabine) ou encore à des protocoles d’investigation. Si le patient ne peut supporter ces tentatives thérapeutiques du fait de son âge ou d’une co-morbidité rédhibitoire, on s’en tiendra à une attitude palliative visant à compenser les conséquences de l’insuffisance médullaire par des transfusions d’érythrocytes en cas d’anémie, de plaquettes en cas de syndrome hémorragique menaçant ou l’usage d’antibiotiques puissants en cas d’infection.
_ Les SMD de faible risque qui représentent donc la majorité des cas posent principalement le problème de l’anémie et des besoins transfusionnels qui en résultent. La décision de transfuser de concentrés érythrocytaires se guide sur la concentration d’hémoglobine mais aussi sur l’âge et les pathologies associées. La transfusion n’est qu’un expédient car les patients qui en sont dépendants restent finalement anémiés le plus clair de leur temps avec un effet délétère des variations de la concentration d’hémoglobine sur leur qualité de vie et leur système cardiovasculaire sans compter la surcharge en fer. Il est d’ailleurs démontré que la dépendance transfusionnelle constitue en soi un facteur pronostique péjoratif indépendant du score IPSS. En dehors du cas particulier du syndrome 5q- qui répond remarquablement au lénalidomide ou Revlimid®, un dérivé du thalidomide, les efforts se sont tournés vers les produits capables de stimuler l’érythropoïèse. Les androgènes ont été longtemps la seule ressource mais la nécessité d’une imprégnation prolongée et les fréquents effets secondaires les ont pratiquement fait abandonner, à l’exclusion du Danatrol souvent bénéfique sur la thrombopénie. Un accord professionnel s’est établi sur l’utilisation des érythropoïétines humaines recombinantes (rHu-EPO) en fonction de la concentration d’EPO sérique endogène. La décision d’utiliser ce traitement et sa prescription initiale sur ordonnance de médicament d’exception relèvent de l’hématologue mais le renouvellement sans modification de posologie est accessible au médecin généraliste jusqu’à concurrence d’une année. En raison du coût de cette thérapeutique (voir tableau ci-dessous), le renouvellement ne saurait concerner que les patients chez qui l’efficacité est avérée par un gain de concentration en hémoglobine de plus de 2g/dl au terme de deux mois de traitement. L’adjonction de facteur de croissance granulocytaire (G-CSF, Neupogen® ou Granocyte®) semble apporter dans certaines études en cours un bénéfice supplémentaire par action synergique.
Le tableau I résume les recommandations des groupes d’experts concernant le traitement de l’anémie dans les SMD de faible risque. Le tableau II donne les principales caractéristiques des érythropoïétines humaines recombinantes disponibles.
Tableau I : traitement de l’anémie des SMD de faible risque
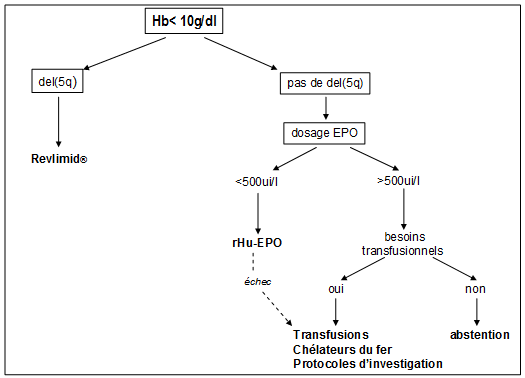
Tableau II : Erythropoïétines humaines recombinantes
|
Spécialité |
EPREX® époïétine alfa |
Néo-RECORMON ® époïetin bêta |
ARANESP ® darbopoïetin alfa |
|
Dose / semaine |
150-300 unités/kg |
10.000-40.000 unités |
150-300 µg ou 500 µg / 3 semaines |
|
Présentation |
Seringues pré-remplies 3.000 à 10.000 ui Flacons de 1000 à 10.000 et 40.000 ui |
Flacons 500 à 30.000 u.i. Cartouches 10, 20 & 60.000 ui |
Seringues pré-remplies 10 à 100 µg, 150, 300 et 500 µg |
|
Administration |
Injection sous-cutanée |
||
|
Prescription |
Ordonnance de médicament d’exception (1ère prescription par médecin hospitalier) renouvelable par le généraliste jusqu’à 1 an sauf modification de dose |
||
|
Délai de réponse |
3 semaines. Evaluation à 2 mois : réponse si & Hb> 2g/dl |
||
|
Principaux effets indésirables |
Céphalées, HTA, évènements thrombo-emboliques, carence martiale, & plaquettes Rares érythroblastopénies |
Céphalées, HTA, évènements thrombo-emboliques, carence martiale, & plaquettes , rash cutané, allergie |
idem + douleur au point d’injection, arthralgies, oedème |
|
Coût mensuel |
420 à 1260 € |
432 à 1296 € |
1300 à 2600 € |
|
AMM ds SMD ? |
NON (mais accord professionnel) |
||
Une hémochromatose complique souvent l’évolution des SMD de faible risque ; la surcharge en fer est présente d’emblée dans les anémies ou cytopénies réfractaires sidéroblastiques. Dans tous les cas elle se majore avec les transfusions puisqu’une transfusion de 4 concentré érythrocytaires apporte 800mg de fer soit 20 à 25% du capital de l’organisme. La ferritine doit être dosée au bilan initial et il existe une corrélation inverse entre sa valeur et la survie ; l’usage des chélateurs du fer est souvent nécessaire pour limiter la surcharge tissulaire et en prévenir les dégâts. Ce traitement doit être mis en œuvre si la ferritine excède 1000 ng/ml ou bien au-delà de 16 concentrés érythrocytaires transfusés.
Le Desféral® en administration sous-cutanée prolongée reste le traitement de première intention, il s’applique à domicile avec l’aide d’infirmières ou bien de prestataires prenant en charge la fourniture du matériel et l’éducation du patient (Société Vitalaire™ par exemple) ; en cas d’inefficacité ou d’intolérance, il est autorisé de recourir à un chélateur oral : les caractéristiques de ces médicaments figurent dans le tableau III.
Tableau III : chélateurs du fer
|
Chélateur |
DESFERAL® Déféroxamine |
FERRIPROX ® Défériprone |
EXJADE ® Deférasirox |
|
Dose mg/kg/j. |
25-60 |
75 |
20-30 |
|
Présentation |
Ampoules 500 mg et 2g |
comprimés 100, 500 mg |
cp 125, 250, 500 mg |
|
Administration |
Injection SC ou IV sur 8 à 12h , 3 à 5 nuits/ semaine |
3 prises orales par jour |
1 prise orale par jour |
|
Demi-vie |
30 minutes |
3 heures |
12 heures |
|
Excrétion |
urinaire + fécale |
urinaire |
fécale |
|
Principaux effets indésirables |
Réactions locales, cataracte, surdité, retard de croissance, allergie |
gastro-intestinaux, neutropénie / agranulocytose, arthralgies, & enzymes hépatiques |
gastro-intestinaux, rash , &créatinine, &transaminases, vision et audition à surveiller |
|
Coût mensuel |
310-420 € + consommable, IDE |
1500 € |
3600 € |
|
AMM ds SMD ? |
Oui |
Non (thalassémies) |
Oui (2ème ligne) |
Pour les autres cytopénies, les moyens thérapeutiques sont plus limités : la thrombopénie peut répondre au danazol (Danatrol®) après plusieurs mois d’administration ; la transfusion de concentrés plaquettaires n’est indiquée qu’en cas de syndrome hémorragique préoccupant : leur efficacité ne dépasse pas 2 à 4 jours et leur répétition entraîne fréquemment une immunisation qui les rend inefficaces. Les nouvelles molécules analogues de la thrombopoïétine (TPO) sont en cours d’évaluation avec des résultats encourageants. La neutropénie peut bénéficier de petites doses de facteur de croissance granulocytaire mais on se limite le plus souvent à traiter les infections par une antibiothérapie à large spectre dès la moindre hyperthermie >38°5.
La transformation leucémique aiguë assombrit le pronostic à brève échéance et ne peut bénéficier que de thérapeutiques palliatives.
De nouveaux traitements font leur apparition : immunomodulateurs et anti-angiogéniques (thalidomide et dérivés), déméthylants et dé-acétylants, tous produits onéreux en cours d’évaluation.
Comme dans nombre de maladies graves du sujet âgé, il est à craindre que l’augmentation des cas et le coût de ces nouveaux traitements ne se heurtent tôt ou tard à des contraintes économiques.
BIBLIOGRAPHIE :