pour la Commission Pédagogique (CP)
Le système CRISP-Cas9.
CRISPR-Cas9, le couteau
suisse de la génétique
Pr Bruno DELOBEL, Centre de Génétique, hôpital Saint Vincent de Paul, GHICL, Lille
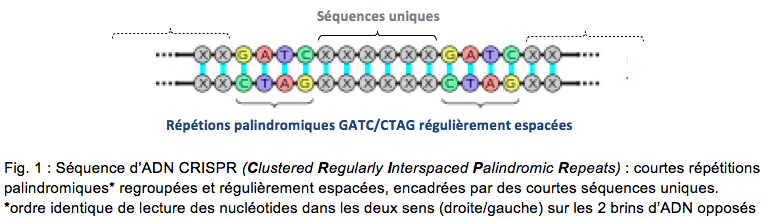
1987 des chercheurs japonais de l’université d’Osaka rapportaient une découverte apparemment mineure [1]. Leurs travaux sur la séquence d’un gène d’enzyme bactérien (phosphatase alcaline d’Escherichia coli) les amenèrent à découvrir un segment d’ADN voisin inhabituel. Celui-ci était constitué de courtes séquences de nucléotides répétés, flanquées de courts segments d’ADN uniques (fig.1). Ils notaient que « la signification biologique de ces séquences étaient inconnues ». Trois décennies plus tard ce qui semblait être une observation anecdotique est utilisé de façon intensive, sous forme d’un système appelé d’un nom quasiment imprononçable : CRISPR-Cas qui révolutionne les techniques de modification du génome. En effet, ce système associant une enzyme protéique coupant l’ADN, Cas, et un ARN dérivé notamment des séquences CRISPR identifiées en 1987 (fig. 1), permet la modification ciblée, simple, efficace et peu onéreuse de la séquence d’un génome. Ceci offre des perspectives dans des domaines aussi variés que la médecine, l’agriculture, la chimie… Pour l’Homme, les recherches sont centrées sur les maladies (héréditaires, cancéreuses voire infectieuses…) avec la possibilité de créer relativement facilement des modèles animaux de pathologies humaines et de redonner une perspective à la thérapie génique, dont les résultats précédents n’avaient pas réalisés les espoirs escomptés. Sa facilité d’utilisation suscite des inquiétudes quant à la possibilité théorique de modifier le génome de tous les organismes permettant des dérives, appelant experts et simples citoyens à un large débat sur le sujet (thérapie ou amélioration génique germinales…).
Historique
Les modifications du
génome
Initialement, jusqu’au début des années 90, les techniques d’obtention d’organismes génétiquement modifiés ne permettaient pas d’introduire la séquence d’un ADN étranger dans une région précise (intégration aléatoire). Ainsi, cet ADN pouvait s’intégrer dans un gène ou une région régulatrice importante, provoquant ainsi une dérégulation de l’expression génique.
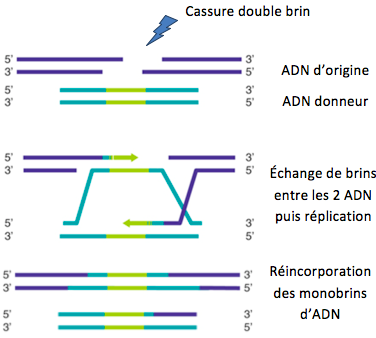
Ce problème crucial de ciblage, a été partiellement résolu grâce à des travaux basés sur la recombinaison homologue (échange entre 2 molécules d'ADN doubles brins identiques ou similaires ; l’un des systèmes de réparation de l’ADN initié par une cassure double-brin de l'ADN) sur les cellules souches embryonnaires de souris (M. Capecchi, M. Evans et O. Smithies, prix Nobel en 2007). Ces travaux permettaient d’introduire un fragment d’ADN à un endroit désiré (fig. 2). Ces cellules recombinées étaient ensuite utilisées chez ces souris portant dans leur génome cette modification, transmissible à leurs descendants. Malgré les bénéfices incontestables de cette technique, celle-ci, lourde et couteuse (près d’un an de travail pour une étude spécifique), était peu efficace en termes de taux de réussite de la recombinaison homologue et pratiquement limitées aux modèles murins.
Vient ensuite dans les années 2010, les nucléases (à doigts de zinc, méganucléases, TALEN…) : véritables « ciseaux moléculaires spécifiques de l’ADN» coupant l’ADN en des endroits précis et ciblés avec une augmentation considérable des évènements de recombinaison facilitant ainsi l’intégration du transgène. Elles apportaient une approche novatrice notamment par l’absence de nécessité de passer par des cellules souches embryonnaires, ouvrant ainsi ces techniques à de nombreux organismes multicellulaires. Cependant, la nécessité pour chaque étude particulière de concevoir des nucléases spécifiques et la variabilité d’efficacité en fonction des organismes, des types cellulaire et des gènes étudiés, rendaient ces techniques relativement lourdes sur le plan pratique.
Et c’est là qu’entre en scène, en 2012, l’outil révolutionnaire de CRISPR-Cas9. Il faut cependant revenir brièvement sur l’historique de cette découverte :
Ce n’est qu’en 2002, quinze ans après l’identification de courtes séquences d’ADN répétés palindromiques d’E. coli évoquées précédemment, que le nom officiel de ces séquences est donné sous la forme d’un acronyme : CRISPR (Clustered Regularly Interspaced Palindromic Repeats : « courtes répétitions palindromiques regroupées et régulièrement espacées »), reflétant leur étrange conformation. En 2005, trois publications de 3 équipes différentes indiquent que les fragments d’ADN qui séparent ces courtes répétions ne correspondent pas à de l’ADN bactérien mais à des séquences de virus qui infectent les bactéries (des bactériophages). En 2007, des chercheurs en biotechnologie d’une entreprise agroalimentaire danoise (Danisco) découvrent que les bactéries alimentaires Streptococcus thermophiles utilisées pour fabriquer les yaourts résistent mieux aux bactériophages quand elles contiennent ces séquences CRISPR. Tout se passe comme si la bactérie gardait en mémoire la trace de la première infection virale en incorporant dans son génome un fragment du virus et qu’elle s’en servait ensuite pour se débarrasser de l’intrus rencontré par elle (ou par ses ancêtres), agissant ainsi comme un « vaccin ».
Un kit moléculaire
précis, simple et universel pour modifier les génomes
En 2012, la française, Emmanuelle Charpentier et l’américaine, Jennifer Doudna publient un article retentissant dans Science [3]. Sur le plan fondamental elles décrivent avec précision le mécanisme par lequel le virus est éliminé par le système CRISPR-Cas9. Sur le plan pratique, elles proposent un kit universel révolutionnaire permettant de modifier à volonté le génome à un endroit précis.
Le système CRISPR-Cas9 est composé d’une endonucléase, la protéine Cas9 (pour CRISPR-associated 9), associée à un ARN guide (fig. 3). Cet ARN est composé d’une séquence fixe se liant à Cas9 et d’une séquence variable permettant la reconnaissance de l’ADN génomique cible. Le positionnement du couple ARN-Cas9 sur leur cible d’ADN permet à la nucléase Cas9 de la couper à un endroit précis (en aval d’une séquence nucléotidique NGG appelée PAM (Proto-spacer Adjacent Motif)).
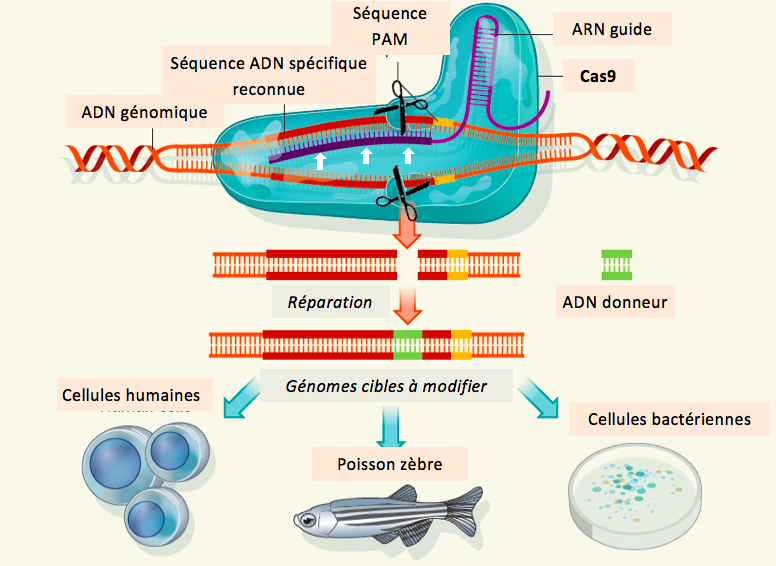
À peine quelques mois après la publication de leur article, plusieurs laboratoires travaillant sur des cellules de mammifères (y compris humaines) montraient la justesse de leur proposition et découvraient que le système CRISPR-Cas9 était incomparablement plus efficace, simple et rapide que les techniques précédentes les plus sophistiquées. Depuis, la technique s’est répandue et les génomes de très nombreux organismes ont été modifiés (riz, tabac, maïs, levures, drosophiles, …).
Un champ immense d’applications
Tel un couteau suisse, l’enzyme Cas9 a été utilisée pour remplir de nombreuses fonctions : modifier les séquences d’un ou de plusieurs gènes simultanément pour les inactiver ou au contraire les réparer. La technique n’a de restriction, pour l’instant, que l’imagination des chercheurs qui l’utilisent.
Les biologistes cherchent également à améliorer la technique, notamment pour éviter les erreurs de ciblage et augmenter son efficacité. Dans cette optique, Cas9 a été remplacée par une autre enzyme bactérienne de fonction similaire, Cpf1. Cette enzyme forme avec l’ARN guide un complexe plus petit qui pénètre plus facilement dans les cellules ou les tissus et qui semble encore plus précis que le complexe CRiSPR-Cas9.
La recherche médicale n’est pas en reste et les exemples d’utilisation de la technique CRISPR/Cas9 sont nombreux. Citons par exemple les travaux effectués chez la souris qui a corrigé une maladie génétique incurable du foie, la tyrosinémie, ou encore, plus récemment, les recherches publiées simultanément par 3 équipes sur la guérison de souris modèles de la myopathie de Duchenne (dégénérescence musculaire qui touche 1 garçon sur 5000). Leurs résultats laissent penser qu’on pourrait utiliser CRISPR-Cas pour « réparer » des patients ainsi affectés. Mais d’importantes améliorations techniques sont nécessaires avant de passer aux essais cliniques.
Jusqu’où peut-on
aller ?
La puissance de l’outil commence à inquiéter. Si l’on admet facilement la possibilité de réparer un gène défectueux chez un individu malade, est-on prêt à accepter de modifier le génome d’un enfant à naître ? Si de telles interventions sont inenvisageables en France, qu’en est-il dans les autres pays ? En avril 2015, une équipe chinoise s’est servie de CRISPR-Cas9 pour modifier le génome d’embryons humains, contrevenant à la convention d’Oviedo ratifiée par 28 pays européens, dont la France en 2011. Les biologistes de l’équipe de Junjiu Huang (université de Sun-Yat-sen, Canton) ont travaillé sur des embryons humains non viables (car porteurs de chromosomes surnuméraires) et ont testé leur capacité à corriger une b-thalassémie. Même si une faible fraction des 80 embryons traités avec le kit CRISPR-Cas9 a été corrigée, nul doute que, la technique s’améliorant constamment, le pourcentage de réussite ira croissant. Modifier le génome d’un embryon humain, c’est par définition modifier le génome de ses futures cellules gamétiques (spermatozoïdes ou ovocytes) et ainsi transmettre la modification apportée à sa descendance potentielle. C’est, comme le formulent déjà certains, la porte ouverte à l’eugénisme.
Les Académies des Sciences de plusieurs pays (Angleterre, Chine, France, USA) se sont réunies à Washington début décembre 2015 pour débattre de l’utilisation biomédicale de CRISPR-Cas9. Elles ont reconnu la puissance et la facilité d’utilisation de cet outil pour traiter un grand nombre de pathologies d’origine génétique ou virale. Mais elles ont aussi noté que des améliorations techniques considérables étaient encore nécessaires avant d’envisager des essais cliniques. Ce qui pose le plus de questions n’est pas tant de modifier le génome des cellules somatiques (cellules musculaires, du sang, du foie…) que de modifier les cellules germinales et, partant, la descendance à venir et ainsi orienter l’évolution de l’espèce humaine. Alors il est temps que non seulement les experts scientifiques et les spécialistes médicaux mais aussi l’ensemble de la société s’emparent de la réflexion, débattent et participent aux décisions.
1 Ishino Y, Shinagawa H,
Makino K, Amemura M, Nakata A. 1987 Nucleotide sequence of the iap gene,
responsible for alkaline phosphatase isozyme conversion in Escherichia coli,
and identification of the gene product. J. Bacteriol. 169, 5429–5433.
2 Charpentier
E, Doudna JA. 2013 Biotechnology: Rewriting a genome. Nature.
495, 50-51.
3 Jinek M,
Chylinski K, Fonfara I,Hauer M, Doudna J A., Charpentier E. 2012 Programmable
Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science.
337:816-21